
Our paper investigating how pathogen richness, the cost of expressing MHC variants, and pathogens' mutation rate shape the individual MHC copy number is out!
17 May 2019MHC genes, which code for proteins responsible for presenting pathogen-derived antigens to the host immune system, show huge copy-number variation both between and within species. However, the evolutionary forces driving this variation are not understood very well. We used computer simulations to investigate whether the evolution of the number of MHC variants in the genome can be shaped by the number of pathogen species the host population encounters (pathogen richness).
The majority of MHC research has focused on amino acid sequence polymorphism. However, an aspect of MHC diversity that has received less attention is the number of MHC variants carried by individuals. Expressing many MHC molecules could ensure that an individual could present antigens of most pathogen species encountered, but this comes at a cost, such as enhanced negative selection on lymphocytes leading to holes in T-cell receptor repertoire. The observed number of MHC variants carried by individuals is typically much lower than the number found in the population. For example, in humans, there are 6–7 classical MHC loci, allowing for up to 12–14 different variants in a fully heterozygous individual. In contrast, the number of currently identified MHC alleles summed across those loci in the human population exceeds 17,000 (IPD-IMGT/HLA Database, Release 3.30.0).
We were using computer simulations based on a framework that has previously been shown to be effective in recovering some of the essential features of MHC evolution, such as high polymorphism, frequency-dependent selection, heterozygote advantage and positive selection. Our model simulated interactions of MHC molecules and antigens produced by pathogens by matching strings of bits, which can mutate both in hosts and in parasites.
Our simulations showed that evolution of the number of MHC genes in the genome is driven by a complex interaction between three factors we explored: pathogen richness, the intrinsic cost of expressing additional MHC variants, and pathogen mutation rate. In contrast to verbal arguments, our results indicate that pathogen richness does not always select for MHC gene family expansion.
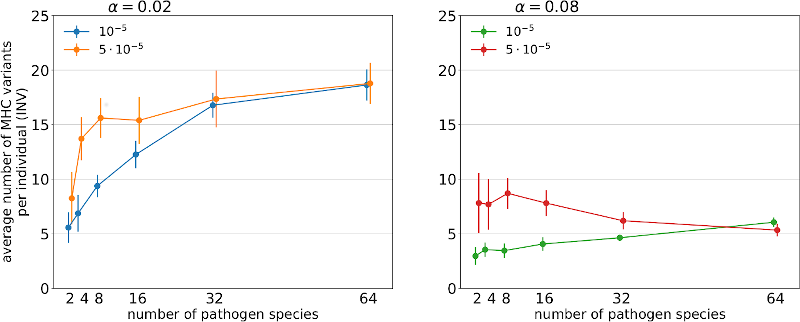 Fig.1. Relationship between the number of pathogen species and the average numbers of unique MHC variants present in a genome of a host under two penalty factors (α = 0.02; 0.08 –panels) and two pathogen mutation rates.
Fig.1. Relationship between the number of pathogen species and the average numbers of unique MHC variants present in a genome of a host under two penalty factors (α = 0.02; 0.08 –panels) and two pathogen mutation rates.
The paper was published in the PLOS Computational Biology.
Cite:
The code of this project can be found at my GitHub repository.